The Protein Mystery: How AlphaFold2 Solved Biology's Toughest Puzzle
Updated July 10, 2025
Introduction
Background
Methodology
CASP-14
The AlphaFold2 Algorithm
Data Inputs
- Target sequence: Amino acid sequence of the protein to be folded.
- Multiple Sequence Alignment (MSA): Similar amino acid sequences from various organisms, in a 3D matrix.
- Templates: Known (folded) structures of homologous proteins (proteins similar to the target sequence).
AlphaFold2 Network
-
Preprocessing: The target sequence is used to search genomic databases, to generate the MSA and find homologous templates. These construct the MSA and pair representation.
A pair representation is a 2D matrix containing information about the relationship between any two residues (residues are amino acids in a protein that are linked in a chain).
- Evoformer Module: Enriches the MSA and pair representation through 48 iterations, where the output of one iteration is the input for the next iteration. It uses two machine learning architectures: transformer and attention.
- Structure Module: Uses the enriched MSA and pair representation to build and refine the predicted 3D structure of the protein in 8 iterations. The output of one iteration is the input for the next iteration. Also, attention mechanism was creatively applied to 3D space here.
Evaluation Metrics
- Global Distance Test - Total Score (GDT_TS): Estimates how many amino acids in the prediction are where they should be.
- Local Distance Difference Test (lDDT): Checks if distances between nearby atoms are as they should be.
- Template Modeling Score: Tells how similar a prediction is to the original structure.
- Root Mean Square Deviation (RMSD): Measures the average amount of error in amino acid positions.
Results
Superior Accuracy
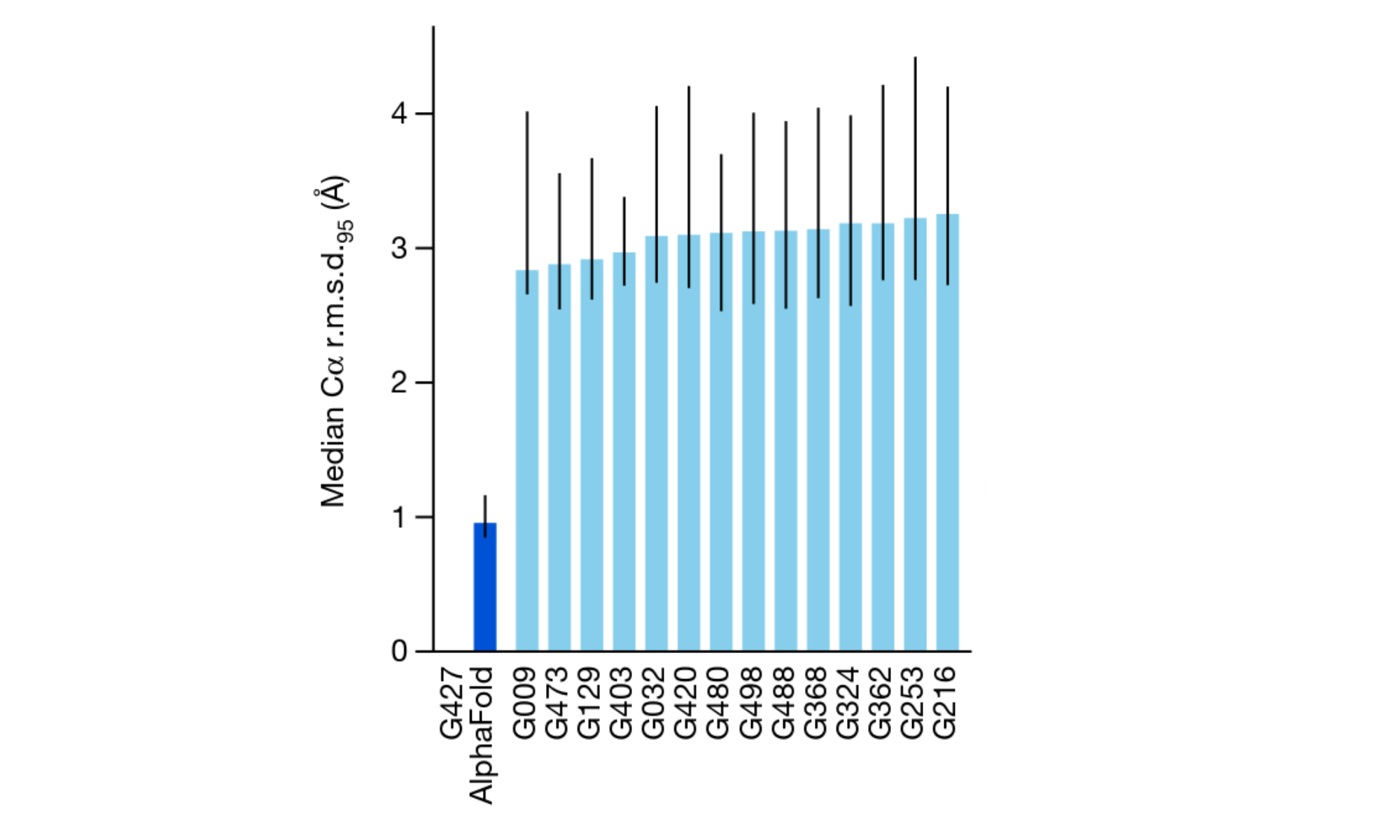
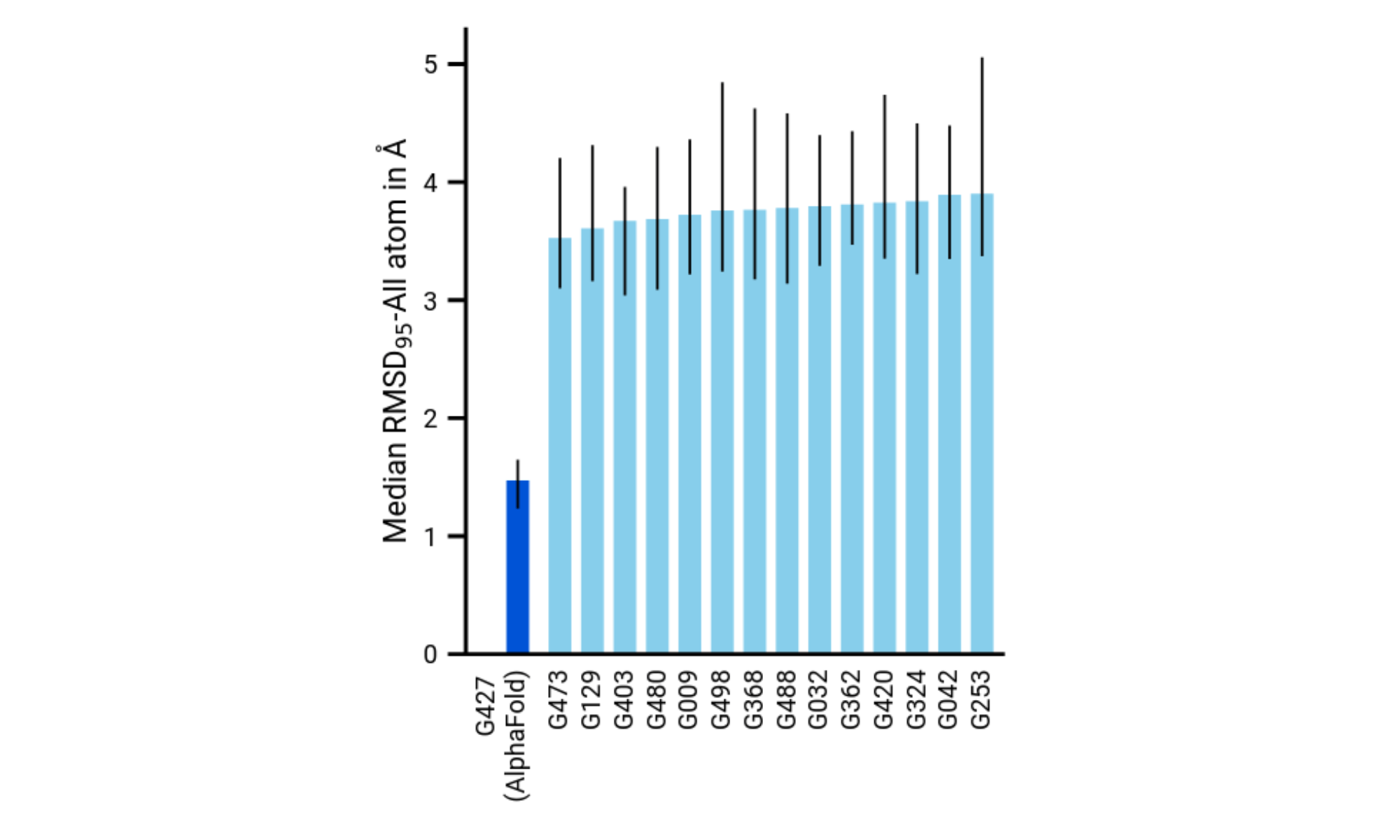
Reliable and Transferable
Scalable and Novel
Discussion
Implications
- Narrowed sequence-structure gap: Out of over 200 million known protein sequences, only the structure of about 200,000 proteins had been determined. AlphaFold narrowed this huge gap by predicting over 200 million protein structures.
- Advanced biological understanding: AlphaFold’s reliable protein structures give bioinformaticians the ability to understand how proteins function at an atomic level, thereby deepening our understanding of how many diseases start and progress.
- Personalized drug discovery: With AlphaFold, researchers can predict how specific genetic mutations in each patient’s protein makeup can negatively alter protein structures, and they can proactively design new drugs to overcome them. Being able to reliably predict the structure of viral and bacterial proteins can also be used to design stronger and precise vaccines.
Limitations
- Protein Multimers: AlphaFold2 was trained on single protein molecules, so it struggled to predict structures for multimers (two or more proteins bound together as one unit).
- Intrinsically Disordered Regions (IDRs): IDRs are unstable parts of proteins that don't have a fixed 3D structure. Since AlphaFold2 predicts a single, static 3D structure, it gives low-confidence predictions for IDRs.
- Structural Changes: Proteins need to move and change shape to do their jobs. AlphaFold2 takes a snapshot of a protein in one shape, but it doesn't capture the full range of shapes that a protein can take.
Future Directions
- Mutations: AlphaFold2 can't predict the consequences of mutations to a protein's structure. This is very crucial to understanding diseases and drug resistance, so it’s the next frontier.
- Generative AI: With Gen AI models, we can design protein sequences that fold into desired structures with specific functions. This will be very useful for drug design and even industrial use.
- Enhanced Algorithms: Solving the limitation of structural changes or IDRs might involve combining AlphaFold with other computational methods like molecular dynamics simulations.
Reflection
Conclusion
References
- Jumper, J., Evans, R., Pritzel, A., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2